CRISPR Kullanan İlk Tedaviyi, Orak Hücre Hastalığı ve Talasemi için İngiltere'de Onaylandı
Tıp adına heyecan uyandıran bir gelişme, Birleşik Krallık'ta gerçekleşti: orak hücreli anemi ve beta talasemi gibi genetik kan bozukluklarının tedavisi için dünyada ilk kez CRISPR kullanan bir gen düzenleme tedavisi onaylandı.
Bu onay, 12 yaşından büyük bireyler için CRISPR teknolojisinin kullanımını mümkün kılarak tıbbi bir dönüm noktasını işaret ediyor. Bu teknoloji, genetik materyali hassas bir şekilde kesip biçme yeteneğine sahip olup, bu özelliği ile Nobel ödülüne layık görülmüştür. Tedavi sürecinde, hastalardan alınan kök hücreler, CRISPR kullanılarak laboratuvar ortamında özel olarak düzenlenir ve daha sonra bu hücreler hastaya geri verilir. Bu sayede, hastaların orak hücre hastalığı ve beta-talasemi gibi genetik kan bozukluklarının olumsuz etkilerinden en az 12 ay süreyle arındıkları gösterildi.
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) teknolojisi, aslında bakterilerin virüslere (bakteriyofajlara) karşı savunma mekanizmalarından evrimleşmiş bir sistemdir. Bakteriler, virüsler tarafından enfekte edildiklerinde, CRISPR-Cas9 sistemi kullanarak virüs DNA'sını tanır ve keser. Bu sayede, eğer gelecekte aynı virüs tekrar saldırırsa, bakteri hızlı bir şekilde virüsün DNA'sını tanıyıp yok edebilir. Bu doğal savunma mekanizması, modern gen düzenleme araçları geliştirmek için bir model olarak kullanılmıştır. Yani, CRISPR-Cas9 sistemi, bakterilerin virüslere karşı geliştirdiği bir savunma mekanizmasından türetilmiştir.
Birleşik Krallık'ın ilaç ve sağlık ürünleri düzenleyici kurumu (MHRA), CRISPR adı verilen gen düzenleme aracını kullanan Casgevy isimli ilacın kullanımını 15 Kasım 2023'te onayladı. Gen düzenleme teknolojisi olan CRISPR, 2020'de Nobel ödülü kazandırmıştı.
Beta Talasemi Nedir?
Beta-talasemi, beta-globin alt birimindeki mutasyonlar yoluyla kandaki normal hemoglobin ve kırmızı kan hücrelerinin azalmasına neden olan ve vücuda yetersiz oksijen verilmesine yol açan bir tür kalıtsal kan hastalığıdır.
Beta-talasemi, tıpta "anemi" dediğimiz kansızlığın nedenlerinden biridir. Azalmış kırmızı kan hücresi seviyeleri, baş dönmesi, halsizlik, yorgunluk, kemik anormallikleri ve daha ciddi komplikasyonlar gibi bir dizi sağlık sorununa yol açabilir.
Beta talasemi anne ve babadan çocuklara kalıtsal olarak geçen, önlenebilir bir kan hastalığıdır. Türkiye’nin de içinde olduğu Akdeniz ülkelerinde önemli bir halk sağlığı sorunudur. Taşıyıcıların saptanması, genetik danışma ve doğum öncesi tanı konabilmesiyle engellenebilir bir hastalık olmasına rağmen, dünyada her yıl en az 365.000 talasemi hastası doğmakta ve tedavi görmektedir. Türkiye’de yaklaşık 1.300.000 talasemi taşıyıcısı ve 4.500 kadar talasemi hastası vardır.
Beta talasemi hastalığı ağır, tedavi düzgün sürdürülmezse yaşam süresini belirgin kısaltan ve yaşam kalitesini çok olumsuz etkileyen bir hastalıktır. Hastalığın tedavisi zordur ve maliyeti çok yüksektir. Talasemili bir hastanın yıllık tedavi maliyeti 10 bin dolar civarındadır. Bu nedenle, hastalıklı bireylerin doğmasını engellemek çok önemlidir ve gerekli koruyucu önlemlerin alınması devlet tarafından da desteklenmektedir.
Beta-talaseminin en şiddetli şekli olan transfüzyona bağımlı beta-talasemi, standart tedavi yöntemi olarak genellikle yaşam boyu kırmızı kan hücresi transfüzyonlarını gerektirir. Bu düzenli transfüzyonlar, vücutta aşırı demir birikmesi nedeniyle kalp, karaciğer ve diğer organlardaki problemler de dahil olmak üzere, kendi başına birçok sağlık komplikasyonuyla ilişkilendirilebilir.
İlgili konu: Beta-Talasemi Tedavisinde İlk Hücre Bazlı Gen Terapisi Ağustos 2022'de FDA Onayı Aldı
Orak Hücreli Anemi Nedir?
Orak hücreli anemi (tıbben "orak hücre hastalığı" daha doğru bir kullanımdır), beta globin geninde meydana gelen mutasyonlardan kaynaklanan kalıtsal kan hastalığıdır. Beta globin genindeki mutasyonlar, anormal hemoglobin üretimine neden olmaktadır. Anormal hemoglobin kırmızı kan hücrelerinin katılaşmasına, anormal (orak) şekil almasına ve kan damarlarını bloke etmesine sebep olur. Orak hücreli anemiye sahip hastalarda akut ağrılar, akut göğüs sendromları, organ hasarları görülür. Tüm bu durumlar, hastalarda yaşam süresini kısaltır.
Casgevy'nin Onay Süreci
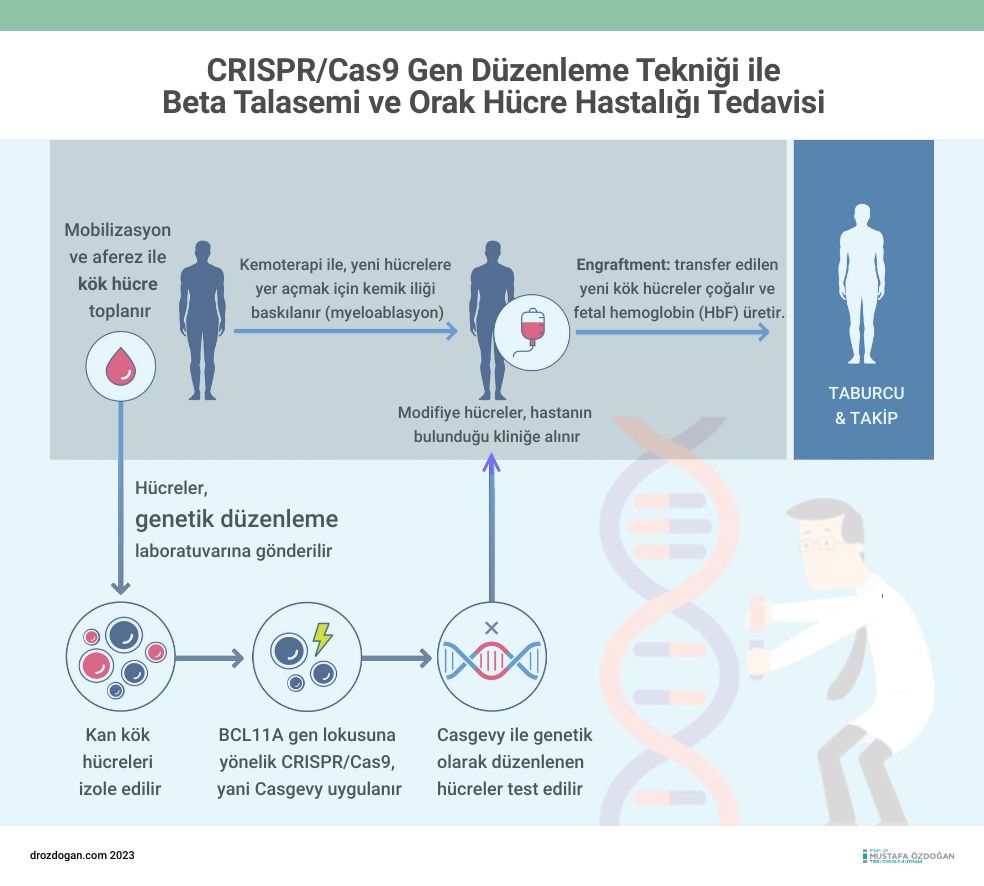
Klinik deneylerde, Casgevy'nin çoğu katılımcıda sağlıklı hemoglobin üretimini yeniden sağladığı, böylece hastalık belirtilerini hafiflettiği tespit edildi. Ayrıca, tedavi sırasında ciddi bir güvenlik endişesi tespit edilmedi ve ilacın güvenliği yakından takip ediliyor. İlaç, hastanın kemik iliğinden alınan kök hücreleri laboratuvarda genetik olarak düzenleyerek ve sonrasında hastaya geri enjekte edilerek uygulanıyor.
Bu tedavi, Vertex Pharmaceuticals ve CRISPR Therapeutics tarafından geliştirilmiş ve Casgevy markası altında ticarileştirilmiş bir gen düzenleme ilacıdır.
Çalışma Mekanizması Nedir?
Tek sefer uygulanan bu tedavi, hasta kişinin kemik iliğinden alınan kök hücrelerdeki BCL11A gen lokusunu CRISPR teknolojisi ile düzenleyerek, yüksek seviyelerde fetal hemoglobin (HbF) üretimini artırır. Fetal hemoglobin, orak hücre hastalığı ve beta talasemisi olan hastalarda anormal yetişkin hemoglobin (HbA) yerine oksijen taşımada rol alır. Bu yolla, hastalığın şiddeti azaltılabilir ve belirtiler üzerinde olumlu etkiler sağlanabilir.
Neden BCL11A Geni Seçildi?
BCL11A geni, yetişkin eritrositlerdeki gama (γ)-globin ifadesini ve fetal hemoglobin (HbF) üretimini baskılayan bir gen olarak tanımlanmıştır. Orak hücre hastalığı (OHH) ve beta talasemide (β-talasemi) HbF üretimini artırarak hastalığın şiddetini azaltmak için terapötik stratejiler geliştirilmektedir. BCL11A'nın baskılanması, özellikle OHH'de HbF seviyelerini artırır ve hücrelerin orak şeklini almasına neden olan hemoglobin şeklinin bozulmasını azaltır, bu nedenle OHH için gen tedavisinde en iyi yaklaşım olarak kabul edilmektedir.
BCL11A'nın down-regülasyonu, yani aktivitesinin azaltılması, HbF'nin endüksiyonu için umut verici bir terapötik strateji olarak görülmekte ve bu yöntem OHH ve β-talasemi hastaları için uygulanmaktadır. Fetal hemoglobin, yetişkinlikte devam eden HbF seviyelerindeki farklılıklar, OHH ve β-talasemi sendromlarının şiddetini etkileyebilmektedir. BCL11A, HbF'nin baskılanmasında kritik bir düzenleyici olarak tanımlanmıştır ve bu genin patojenik varyantları, HbF üretiminin artırılması ve böylece hastalığın şiddetinin hafifletilmesi konusunda fikirler sunmuştur.
Hasarlı Geni Düzeltmek Yerine Kurnazca Bir Çözüm
CRISPR teknolojisi genellikle genetik yapıyı doğrudan düzenlemek için kullanılır, ancak orak hücre anemisi ve beta-talasemi gibi bazı durumlarda, hasarlı geni düzeltmek yerine farklı bir strateji uygulanmaktadır. Bu hastalıklarda problem, anormal veya eksik beta globin proteininin üretimi sonucu bozuk hemoglobin yapımıdır. CRISPR teknolojisi ise BCL11A genini hedef alarak, beta globin yerine yüksek seviyelerde fetal hemoglobin (HbF) üretimini teşvik eder.
Bu yaklaşım, hasarlı veya eksik proteinleri doğrudan düzeltemeyen ancak başka yollarla tedavi etmeye çalışan bir "kurnazlık" veya dolaylı bir strateji örneğidir. Bununla birlikte, tüm genetik hastalıklarda böyle kurnazca çözümler bulmak her zaman mümkün değildir. Özellikle kanser gibi kompleks hastalıklarda, genetik değişimler çok çeşitli olabilir ve temel problemi hedef almak yerine alternatif yolları aramak her zaman istenen sonucu vermeyebilir. Kanser tedavisinde, genellikle birçok farklı genetik ve moleküler hedefe yönelik çok yönlü yaklaşımlar gereklidir. Ancak, CRISPR teknolojisinin gelişmesi ve daha fazla araştırma ile birçok genetik hastalıkta yeni tedavi yöntemleri geliştirme potansiyeli vardır.
Klinik Çalışma Sonuçları
Casgevy'nin onayı, orak hücre hastalığı olan 45 hastayı kapsayan ve bu hastaların 29'u üzerinde yapılan bir çalışmanın sonuçlarına dayanıyor. Bu hastaların hemen hemen hepsi, tedaviden en az 12 ay sonra ciddi ağrı krizlerinden muaftı.
Beta talasemi denemesinde, 54 hastanın 42'si klinik denemede ana etkinlik ara analizi için yeterince uzun süre kaldı. Bu uygun hastaların 39'u tedaviden en az 12 ay sonra kırmızı kan hücresi transfüzyonuna ihtiyaç duymadı. Kalan üç hasta, transfüzyon ihtiyacında %70'ten fazla azalma gösterdi.
Denemelerde bildirilen yan etkiler arasında mide bulantısı, yorgunluk, ateş ve yüksek enfeksiyon riski vardı. Bu sorunlar, diğer otolog kök hücre nakli işlemlerine benzer problemlerdir. Çalışmalardan ciddi yan etkiler bildirilmemiştir.
Casgevy'nin MHRA kararı, gerçek dünya verilerinin ve pazar sonrası yapılan çalışmaların terapinin kullanımını desteklemeye devam etmesi koşuluyla her yıl yenilenebilen bir yıllık geçici pazarlama iznidir.
Bu onay, gen düzenleme teknolojisinin tıbbi tedavilerdeki potansiyelini göstermektedir ve gelecekte bu tür terapilerin daha geniş bir yelpazede kullanımı için kapıları açabilir.
1. U.K. Is First to Approve a CRISPR-Based Therapy, Covering Two Blood Disorders. 16 Nov 2023. https://medcitynews.com/2023/11/u-k-is-first-to-approve-a-crispr-based-therapy-covering-two-blood-disorders/
2. Asmamaw M, Zawdie B. Mechanism and Applications of CRISPR/Cas-9-Mediated Genome Editing. Biologics. 2021 Aug 21;15:353-361. doi: 10.2147/BTT.S326422. PMID: 34456559; PMCID: PMC8388126.
3. Erica B. Esrick, Maureen Achebe, Myriam Armant, et al. Validation of BCL11A As Therapeutic Target in Sickle Cell Disease: Results from the Adult Cohort of a Pilot/Feasibility Gene Therapy Trial Inducing Sustained Expression of Fetal Hemoglobin Using Post-Transcriptional Gene Silencing. Blood 2019; 134 (Supplement_2): LBA–5. doi: https://doi.org/10.1182/blood-2019-132745



