Adrenokortikal Karsinom (Böbrek Üstü Bezi Kanseri) Nedir? Tedavisi Nasıldır?
Adrenal bezler, böbreklerin üstünde bulunan endokrin (hormonal) bezlerdir ve kortizol, aldosteron ve cinsiyet hormonları gibi önemli hormonları üretirler. Adrenokortikal karsinomlar, bu bezlerdeki hücrelerden kaynaklanan nadir kanser türleridir.
Adrenal (böbrek üstü) bezin tek taraflı tümörleri veya kitleleri yaygındır. İşlevsel (hormon salgılayan) veya sessiz ve iyi huylu veya kötü huylu olarak çeşitli sınıflara ayrılırlar.
- Adrenokortikal tümörlerin çoğu, karın görüntüleme çalışmalarında tesadüfen keşfedilen benign (iyi huylu), işlevsiz adenomlardır.
- Bu benign adenomlar hormon salgılayarak Cushing sendromuna, primer aldosteronizme veya çok daha seyrek olarak virilizasyona (kıllanmaya) neden olabilirler.
- Adrenokortikal karsinomlar (böbrek üstü bezi kanserleri), fonksiyonel olabilen ve Cushing sendromuna ve/veya virilizasyona neden olabilen veya belirti vermeyen, karın görüntüleme tetkiklerinde tesadüfen tespit edilen nadir, sıklıkla agresif tümörlerdir.
- Feokromositomalar böbrek üstü bezinin merkezinde bulunan medullanın kromafin hücrelerinden köken alan katekolamin salgılayan tümörlerdir. Benign (iyi huylu) veya malign (kötü huylu, kanserli) olabilirler.
Not: Virilizasyon, kadınların tipik olarak sahip olmayacakları erkeksi fiziksel özelliklerin, erkeklik hormonları olan androjen seviyelerinin yükselmesi sebebi ile ortaya çıkmasıdır.
Aşağıda, böbrek üstü bezlerinin basit anatomisi ve bu küçük organlarla ilişkili hastalıkların kısa bir listesi görülebilir:

Adrenal bezler, böbreklerin üstünde, karın arka bölgesinde, her bir böbreğin üstünde yer alan küçük endokrin bezlerdir. İki ana kısımdan oluşur: korteks ve medulla. Korteks, hormonlar üreten üç farklı tabakadan oluşur: dışta aldosteron, ortada kortizol ve androjenler, içte östrojenler. Medulla, sempatik sinir sistemine bağlı olarak norepinefrin ve epinefrin hormonlarını üretir. Bu hormonlar vücuttaki stres tepkilerinin düzenlenmesinde, kan basıncının kontrolünde, elektrolit dengesinin korunmasında ve diğer pek çok fizyolojik süreçte önemli rol oynarlar.
Adrenokortikal (Adrenal Kortikal) Karsinom Nedir?
Adrenokortikal karsinom (AKK), böbrek üstü bezlerin korteks (kabuk) kısmında başlayan, nadir görülen ve agresif biyolojiye sahip bir kanser türüdür. Bu kanser, adrenal bezlerin salgıladığı hormonlardan biri olan kortizolün üretimini etkileyebilir. AKK, genellikle rastlantısal olarak, yani başka bir sebep için yapılan görüntüleme testlerinde keşfedilir. Patoloji raporlarında adrenal kortikal kanser olarak da adlandırıldığı görülebilir.
Her yıl milyon nüfus başına yaklaşık bir ila iki kişi adrenokortikal karsinom (AKK) tanısı almaktadır. Bununla birlikte, bazı çevresel ve genetik risk faktörlerinin tanımlandığı güney Brezilya'daki çocuklarda sıklığı yaklaşık 10 kat daha yüksektir.
AKK herhangi bir yaşta gelişebilse de hastalığın beş yaşından önce ve yaşamın dördüncü ila beşinci on yılında (40’lı ve 50’li yaşlar) zirve yaptığı bir yaş dağılımı vardır. Genel olarak, hastalığın ilerleme hızı yetişkinlerde çocuklara göre daha hızlıdır.
Kadınlar erkeklerden daha sık AKK geliştirir (1,5 ila 2,5 kat daha fazla).,

Nedenleri
Adrenokortikal karsinomların çoğunluğu spontan görülen hastalıklar olsa da bazıları kalıtsal kanser sendromlarının bir parçası olarak tanımlanabilir. Bu sendromlar arasında Li-Fraumeni sendromu, Beckwith-Wiedemann sendromu ve Multiple Endokrin Neoplazi Tip 1 (MEN1) sayılabilir. Li-Fraumeni sendromu otozomal dominant (baskın) bir hastalıktır ve 17p kromozomundaki TP53 tümör baskılayıcı genindeki mutasyonlarla ilişkilidir. Beckwith-Wiedemann sendromu ise 11p15'teki anormalliklerle ilişkilidir. MEN1 ise 11q kromozomundaki MEN1 genindeki inaktive edici mutasyonlarla ilişkilidir.
Sporadik (kalıtsal olmayan) adrenokortikal karsinomların moleküler mekanizmaları, yukarıda belirtilen çoğu genetik sendromdakiler gibi iyi karakterize edilmemiştir. Ancak, kolorektal kanser için tanımlanan çoklu aşamalı bir tümör ilerleme modeli, benzer bir modelin adrenokortikal karsinomlar için de önerildiği görülmüştür.
TP53 geni, insan kanserlerinde en sık mutasyona uğrayan genlerden biridir. Sporadik adrenokortikal karsinomlarda 17p13'teki lokusta heterozigotluk kaybının sıkça bulunması, TP53 tümör baskılayıcı geninin rolünü düşündürmektedir. Bununla birlikte, 17p13'teki heterozigotluk kaybının sadece yaklaşık üçte biri TP53 mutasyonuyla birlikte görülür. Bu da başka bir henüz tanımlanmamış baskılayıcı genin bu lokusta yer aldığını düşündürür.
Bazı adrenokortikal karsinomlarda, Beckwith-Wiedemann sendromundaki 11p15'teki anormalliklerin de bir rolü olabilir. Ayrıca, CTNNB1 genindeki aktivasyon mutasyonları ve ZNRF3, PRKAR1A, RPL22, TERF2, CCNE1 ve NF1 gibi farklı genlerdeki mutasyonlar da adrenokortikal karsinomlarda gözlemlenmiştir.
Belirtileri
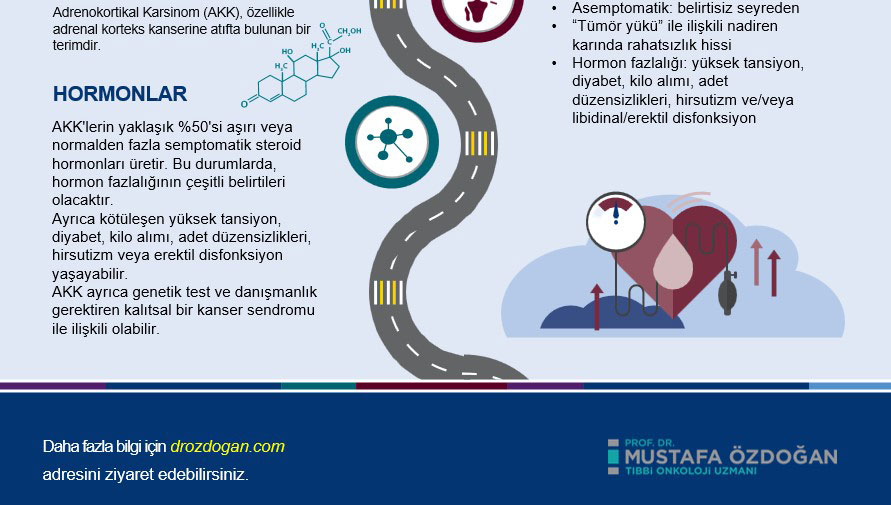
Adrenokortikal kanser (AKK) hastalarının yaklaşık %60'ı hormon fazlalığı sendromu ile kendini gösterir. Hormon salgılayan AKK'lere sahip yetişkinler genellikle sadece Cushing sendromu ( %45) veya hem glukokortikoidlerin hem de androjenlerin aşırı üretimiyle karışık bir Cushing ve virilizasyon sendromu ile karşılaşırlar (%25). Cushing sendromu olan hastaların tedavisi cerrahi veya kemoterapi ile yapıldığında enfeksiyon ve metabolik veya vasküler komplikasyon riski artar.
Glukokortikoid fazlalığı ile ilişkili olan kilo alma, zayıflık ve uykusuzluk gibi klinik belirtiler genellikle çok hızlı bir şekilde gelişir (3-6 ay içinde). Androjenlerin aşırı salgılanması ile birlikte olan hastalar, glukokortikoid fazlalığına bağlı tipik katabolik etkileri (kas ve deri atrofisi) yaşamayabilirler.
AKK belirtilerinin çoğu büyüyen tümörlerden kaynaklanır (karın veya yan ağrısı gibi), veya farklı bir nedenle yapılan radyografik görüntülemede tesadüfen tespit edilen adrenal kitleler ile ortaya çıkarlar.
Tanısı
Hastaların %50'den fazlasında AKK, başka nedenlerle yapılan görüntüleme çalışmaları sırasıdan tesadüfen tespit edilir.
AKK tanısı için hormonal değerlendirmeler yapılması önerilmektedir.
AKK tanısı için yapılan hormonal değerlendirmeler aşağıdakileri içerebilir:
- Serum kortizol düzeyi: Yüksek kortizol düzeyleri Cushing sendromu'na işaret edebilir ve bazı AKK'ların da kortizol salgıladığı bilinmektedir.
- Serum aldosteron ve renin düzeyleri: Yüksek aldosteron ve düşük renin düzeyleri, Conn sendromu olarak bilinen bir hastalığa işaret edebilir ve bazı AKK'lar aldosteron salgılar.
- Serum androjenler: Virilizasyon semptomları olan hastalarda androjen seviyeleri değerlendirilir. AKK'ların bazıları da androjen salgılar.
- Serum östrojen ve progesteron düzeyleri: AKK'ların bazıları östrojen ve progesteron da salgılayabilir.
Bu hormon seviyelerinin yanı sıra, bazı AKK'ların hormon salgılamadığı bilinmektedir. Bu durumda, hormon testleri normal olabilir ve AKK tanısı, tümörün biyopsisi ve histopatolojik incelemesi ile teyit edilebilir.
Bazı radyografik görüntüleme yöntemleri ve özellikle PET-CT taramaları AKK'nin evresini belirlemek için kullanılabilir.
AKK biyopsisi, tümörün konumuna, boyutuna ve hasta durumuna göre farklı yöntemlerle yapılabilir. Bu yöntemler arasında ince iğne aspirasyon biyopsisi (İİAB), tru-cut biyopsi, açık veya kapalı cerrahi biyopsi yer alabilir. İİAB, AKK tanısı için en sık kullanılan yöntemdir ve küçük tümörlerin teşhisinde oldukça yararlıdır. Ancak, büyük tümörlerde ve tümörün tamamının çıkarılması gereken durumlarda açık cerrahi biyopsi veya rezeksiyon tercih edilebilir.
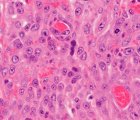
Histopatolojik inceleme, biyopsi örneğinin tanısal değerlendirmesini sağlar. AKK'li hastalarda, tümörün kötü huylu karakterini gösteren belirli histopatolojik özellikler mevcuttur. Bu özellikler arasında yüksek mitotik indeks, hücrelerin nükleer atipi, nekroz ve arteriyel invazyon yer alabilir. Ayrıca, tümörde hormon yapıcı hücrelerin varlığı da tanısal açıdan önemlidir ve bu hücreler, immünohistokimyasal inceleme yöntemleriyle tanınabilirler.
AKK histopatolojik sınıflandırması, tümörün yapısal özelliklerine ve malignitenin derecesine göre belirlenir. En sık kullanılan histopatolojik sınıflandırma yöntemi, Weiss skoru olarak adlandırılır ve tümörün dokusal karakteristiklerine dayanır. Weiss skoru, AKK'nın malignite derecesinin yanı sıra hastalığın prognozunu da tahmin etmekte kullanılır.
Sonuç olarak, AKK biyopsisi, tümörün doğru tanısı ve sınıflandırılması için oldukça önemlidir. Biyopsi yöntemi ve histopatolojik inceleme, tümörün büyüklüğüne, konumuna ve malignite derecesine göre belirlenir. Histopatolojik inceleme sonucunda tümörün malignite derecesi ve prognozu belirlenebilir.
Evreleme
Adrenokortikal kanser için kullanılan farklı evrelendirme / aşamalandırma sistemleri mevcuttur.
Yetişkinler için, 2018 yılında uygulamaya konulmak üzere Amerikan Ortak Kanser Komitesi (AJCC) / Uluslararası Kanser Kontrol Birliği (UICC) tarafından yenilenen 8. tümör, nod, metastaz (TNM) aşamalandırma sistemi kullanılmaktadır. TNM sınıflandırma tanımları, başlangıçta 2004 yılında UICC / Dünya Sağlık Örgütü (DSÖ) tarafından önerilenlerle aynıdır. TNM aşamalandırma sisteminin sekizinci baskısında, önceki 2004 AJCC / UICC sınıflandırmasına göre, evre IV hastalık artık uzak metastazı olan hastalarla sınırlıdır ve lokal invazyon veya bitişik organların invazyonu (T3 veya T4) olan ancak uzak metastazı olmayanlar, evre III hastalığı olan olarak sınıflandırılır. Bu değişiklikler, başlangıçta ENSAT grubu tarafından önerilen değişikliklerle uyumludur.
ENSAT, Adrenokortikal Karsinomda Avrupa Ağı’nın bir parçasıdır ve hastalığın yaygınlığını belirlemek için kullanılan bir sınıflandırma sistemidir.
ENSAT, tümörün boyutu (T), lenf düğümü tutulumu (N) ve uzak metastazların varlığı (M) gibi faktörleri içeren TNM sınıflandırma sisteminin bir varyasyonudur. Aşağıdaki şekilde özetlenmiştir:
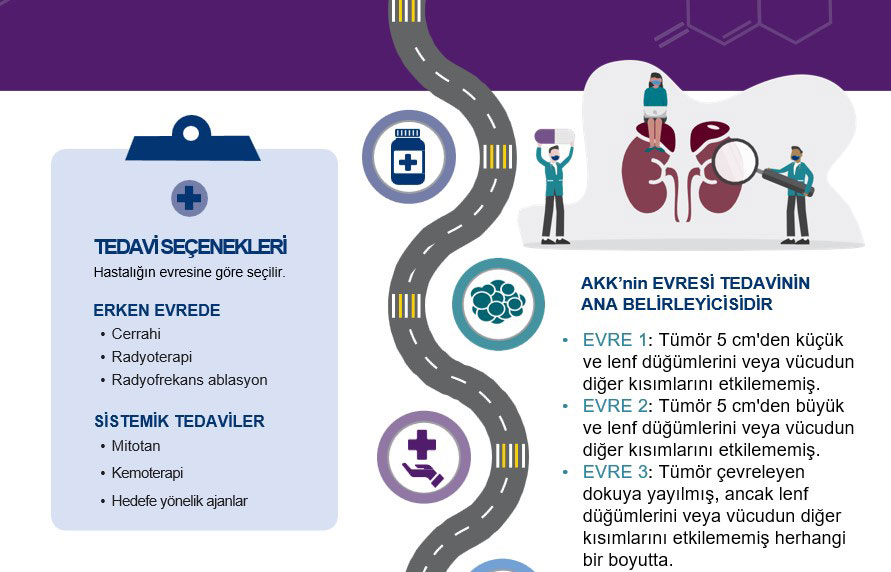
- T1: Tümör 5 cm'den küçük
- T2: Tümör 5 cm'den büyük
- T3: Komşu organlara invazyon (yayılma) var veya invazyon yok ama büyük damarlara tümör teması var
- T4: Uzak organlara veya dokulara invazyon
- N0: Lenf düğümlerinde yayılım yok
- N1: Lenf düğümlerinde yayılım var
- M0: Uzak metastaz yok
- M1: Uzak metastaz var
ENSAT sistemi, TNM sistemi ile birleştirilerek hastalığın evresini belirler. Örneğin, T2N0M0, sadece tümör boyutunun 5 cm'den büyük olduğu durumda, lenf düğümlerine veya uzak organlara metastaz yokluğunda, evre II olarak sınıflandırılır.
Bu sınıflandırma sistemi, doktorların hastalığın gidişatını tahmin etmelerine yardımcı olabilir ve tedavi seçeneklerini belirlemelerine yardımcı olabilir.
ENSAT sınıflandırmasına dayanarak, Almanya’dan bildirilen 416 yetişkin AKK olgusunda, 5 yıllık hastalığa özgül sağkalım oranları aşağıdaki gibidir:
- Evre I – adrenal bezde sınırlı, lokal invazyon veya uzak metastaz olmadan; en büyük tümör boyutu ≤5 cm (T1N0M0): %82
- Evre II – tümör boyutu >5 cm olan ancak risk faktörleri olmayanlarla (T2N0M0) aynı olan evre I: %61
- Evre III – en az biri aşağıdaki faktörlerden birine sahip herhangi bir tümör boyutu: çevre dokularda tümör infiltrasyonu (T3), vena kavada veya renal venlerde tümör trombüsü ve tümör invazyonu (T4), pozitif lenf nodları (N1) ancak uzak metastaz yok: %50
- Evre IV – uzak metastazlar: %13
Çocuklarda evreleme sistemleri yetişkinlerle biraz farklıdır. Bu sistemler, şu üç faktöre göre gruplara ayrılır: tamamen çıkarılmış küçük tümörler (200 g veya daha küçük), tamamen çıkarılmış büyük tümörler (200 g'den büyük) ve kalıntı veya uzak metastatik hastalıklar.
Tedavisi
AKK'nın tedavisi, tümörün tam bir şekilde cerrahi olarak çıkarılmasıdır. Potansiyel olarak cerrahi olarak çıkarılabilir evre I-III hastalar için, tam cerrahi rezeksiyon (tümörü çıkarma), başlangıç tedavisi olarak önerilmektedir. Cerrahi işlem öncesi, tüm hastaların tümörün salgısal aktivitesini belirlemek için tam hormonal değerlendirmeye tabi tutulması gerekmektedir. Cerrahi işlem özellikle bir kortizol üretici tümörü olan hastalar için önemlidir. Bu hastalar, hafif hiperkortizolizm olsa bile, bir dereceye kadar hipotalamik-hipofiz-böbrek üstü ekseni (HPA) baskılanması ve postoperatif adrenal yetersizliği önlemek için glukokortikoid koruması gerektirirler. Cerrahi işlem uzmanlaşmış bir referans merkezinde cerrahlar tarafından yapılmalıdır.
Cerrahi olarak çıkarılabilir tümörlere sahip hastalar için, cerrahi işlem sırasında tümör yayılmasının önlenmesi için cerrahlar tarafından uzmanlaşmış bir ekibin bulunması ve tüm tümörün çıkarılmasının sağlanması gerekmektedir. Cerrahi işlem yapılmadan önce, tüm tümörün hormonal değerlendirmesi yapılmalıdır. AKK'nın yayılmasının en önemli yolu lenfatik drenaj yoluyla olduğundan, şüpheli lenf düğümleri çıkarılmalıdır.
Ameliyat Sonrası Tedavi
Adjuvan (ameliyat sonrası koruyucu) sistemik tedaviye yaklaşım – Evre I ila III hastalığı olan çoğu hastada rezeksiyon teknik olarak mümkün olsa da muhtemelen ilk başvuru anında gizli mikrometastazlar mevcut olduğundan, pek çoğu için küratif (tam şifayı sağlayıcı) değildir. Tamamen rezeke edilmiş AKK'li tüm hastalar için adjuvan tedaviye yaklaşımımız, hastalığın tekrarlama riskine dayanmaktadır:
- Düşük nüks riski – Tam rezeksiyondan sonra nüks (tekrarlama) riski düşük olan çoğu hasta için (evre I ila III, mikroskobik olarak tam [R0] rezeksiyon, Ki-67 ≤ yüzde 10), ilave olarak adjuvan mitotan yerine gözlem öneriyoruz. Bu grup hasta için adjuvan mitotan kullanımı, randomize bir denemede nükssüz hayatta kalma veya genel hayatta kalmada istatistiksel olarak anlamlı gelişmeler göstermedi. Düşük riskli hastalık kriterlerini karşılayan ancak aynı zamanda potansiyel bir nüks riski düşündüren bazı özellikler barındıran hastalar için adjuvan mitotan önerilir. Bu durum için örnekler, evre III düşük dereceli hastalığı olan hastaları veya sınırda mitoz hızı veya Ki-67 skoru olan büyük (>20 cm) evre II tümörü olan hastaları içerir.
- Yüksek nüks riski – Tam rezeksiyondan sonra hastalık nüksü riski yüksek olanlar için gözlem yerine tek başına adjuvan mitotan tedavisi (kemoterapi olmadan) önerilir. Yüksek hastalık tekrarlama riskini gösteren klinik özellikler arasında yüksek dereceli hastalık (Ki-67 > yüzde 10 ve < yüzde 20 veya 50 büyük büyütme alanı başına 20'den büyük mitotik hız); eksik rezeke edilmiş hastalık; intraoperatif tümör dökülmesi veya kırılması; ve vasküler veya kapsüler invazyonu olan büyük, düşük dereceli tümörler.
- Çok yüksek nüks riski – Erken nüks riski çok yüksek olan seçilmiş hastalar için (örneğin, çok yüksek Ki-67 boyanması (≥ yüzde 20) ve yaygın vasküler invazyon/vena kava trombüsü), tipik olarak sisplatin bazlı kemoterapinin eklenmesini önerilir.
Adjuvan radyasyon tedavisi endikasyonları – Tam olarak rezeke edilmemiş AKK'li tüm hastalara, diğer yüksek nüks riski taşıyan evre III hastalığı olanlara, rezeksiyon sırasında tümör döküntüsü olanlara ve yüksek dereceli AKK'li hastalara (50 HPF başına >20 mitotik rakam) adjuvan radyasyon tedavisinin (RT) eklenmesini önerilir.
Mitotan Nedir? Etki Mekanizması Nasıldır?
Mitotan, adrenokortikal karsinom (AKK) tedavisinde kullanılan bir ilaçtır ve en etkili seçeneklerden biridir. Bu ilaç, AKK'nin büyümesini engelleyerek ve kanser hücrelerinin ürettiği hormon seviyelerini düşürerek etki eder.
AKK'li hastalarda, tümörler genellikle kortizol gibi hormonlar üretir ve bu durum, bazı belirti-şikayetlerin ortaya çıkmasına neden olabilir. Mitotan, kortizol üretimini baskılayarak ilişkili belirtileri hafifletir. Mitotan, aynı zamanda tümörün büyümesini yavaşlatarak veya durdurarak hastalığın ilerlemesini önleyebilir.
Mitotan, tedavi sırasında sıklıkla kullanılan bir ilaçtır. İlk olarak, hastalığın şiddeti ve tümörün boyutuna göre bir dozaj belirlenir. Tedavinin ilk aşamasında, hastaya daha yüksek dozlarda verilir ve ardından doz azaltılır. İlacın kullanımı sırasında hastaların düzenli olarak takip edilmesi gerekmektedir. Mitotan, bazı yan etkileri nedeniyle dikkatli bir şekilde kullanılmalıdır. Bu yan etkiler arasında uyku hali, depresyon, karaciğer enzimlerinde artış ve tiroid fonksiyonunda azalma yer alır.
Adjuvan mitotan süresi ne olmalı? Adjuvan mitotan tedavisinin optimal süresi belirlenmemiştir ve hastalığın tekrarlama riskine, toleransa ve hastanın hedef terapötik mitotan seviyelerine ulaştığı maksimum süreye bağlıdır. Rezeke edilmiş düşük riskli AKK'li çoğu hasta için gözlem tercih edilir. Adjuvan tedavi endikasyonu olanlar için en az iki yıl mitotan hedeflenir.
- Rezeke edilmiş yüksek riskli AKK'li hastaları üç ila beş yıllık adjuvan tedavi ile tedavi edilir.
Hedef mitotan seviyeleri – Adjuvan mitotan kullanım dozu, AKK hastaları için farklı olabilir. Genellikle, önerilen başlangıç dozu günde 2-4 gramdır. Mümkünse, terapötik seviyelere ulaşmak ve toksisiteyi önlemek için mitotan tedavisi sırasında kan mitotan seviyesi takip edilerek kullanılmalıdır. Mitotan seviyeleri 14 ve 20 mcg/mL arasında tutulmalıdır.
Adrenal yetmezlik için glukokortikoid replasmanı – Kortizol salgılayan AKK'nin rezeksiyonundan sonra glukokortikoid replasmanı gereklidir. Adjuvan mitotan tedavisi başlatıldığında, kortizol salgılamayan tümörleri olan hastalarda da başlanmalıdır çünkü ilacın adrenolitik aktivitesi çoğu hastada adrenal yetmezliğe yol açacaktır. Glukokortikoid yerine koyma için deksametazon yerine hidrokortizon kullanılır. Mitotan, P450 sistemini indükleyerek kortizol metabolizmasını aşamalı olarak hızlandırdığından, suprafizyolojik dozlar endike hale gelir.
Aldosteron eksikliğinin tedavisi – Mitotan, sonunda aldosteron eksikliğine de neden olabilir. Her ziyarette kan basıncını, her üç ayda bir serum potasyumunu ve altı ayda bir plazma reninini izlemenizi öneririz. Aldosteron eksikliğinin klinik ve biyokimyasal belirtileri ortaya çıktığında, fludrokortizon (günde 0.1 ila 0.3 mg) ilavesi başlatılır ve normal klinik ve biyokimyasal parametreleri eski haline getirmek için ayarlanır.
Tedavi Sonrası Gözetim
Görüntüleme – Tedavi sonrası gözetim, iki yıl boyunca her üç ayda bir, ardından beş yıl boyunca altı ayda bir göğüs ve karın bilgisayarlı tomografi (BT) taramalarını içerir. Tedavi sonrası takip stratejisinde florodeoksiglukoz (FDG)-pozitron emisyon tomografisinin (PET) faydası tartışmalıdır ve uygulama değişkendir.
Biyokimyasal izleme – Tamamen rezeke edilmiş, steroid üreten AKK'li hastalar için, bazı gruplar hastalar iki yıl boyunca üç ayda bir kortizol (hidrokortizon dozundan önceki sabah ölçülür), dehidroepiandrosteron sülfat (DHEAS), androstenedion gibi steroid tümör belirteçleri ile izlenir. İlk tümördeki steroid profiline göre testosteron, östradiol veya mineralokortikoid takip edilebilir. Diğer gruplar, tekrarlayan hastalık kanıtı olmadıkça hormon fazlalığı için rutin olarak taranmazlar.
İleri Evre AKK Tedavisi
Rezeke edilemeyen, tekrarlayan veya ilerlemiş hastalık – Cerrahi olarak çıkarılabilen izole, bölgesel olarak tekrarlayan AKK'li hastalar için, tam cerrahi rezeksiyon ve ardından mitotan tedavisi önerilir. Sınırlı, potansiyel olarak rezeke edilebilir hepatik veya akciğer metastazları olan nadir hastalarda ve kontrol edilemeyen belirtileri ve hormon fazlalığı olan seçilmiş hastalarda da ameliyat düşünülebilir.
- Rezeke edilemeyen hastalığı olan çoğu hasta için, tek başına mitotan veya kemoterapi yerine kombine mitotan artı kemoterapi önerilir. Bununla birlikte, tekli mitotan tedavisi, düşük dereceli, yavaş ilerleyen sınırlı hastalık yükü olan hastalar için makul bir seçenektir.
- Kombine tedavi alan hastalar için etoposid, doksorubisin ve sisplatin (EDP) ile kombinasyon halinde mitotan önerilir.
- Radyofrekans ablasyonu için endikasyonlar – Perkütan radyofrekans ablasyonu (RFA), özellikle tümör çapı <5 cm olanlar için, rezeke edilemeyen bir primer tümörün kısa süreli lokal kontrolünü sağlayabilir.
- Hormon fazlalığının tıbbi tedavisi – Hiperkortizolizmi olan hastalarda (mitotan almayan veya hiperkortizolizmi mitotan tarafından kontrol edilemeyen) veya gerekirse hiperkortizolizmin kontrolünü sağlamak için ketokonazol ve mitotan ile kombine kullanımını önerilir. Glikoz, potasyum seviyeleri, kan basıncı ve enfeksiyonların uygun kontrolü de önemlidir.
- Palyatif RT endikasyonları – Lokal ileri veya uzak metastatik hastalıktan (örn. kemik metastazı) kaynaklanan belirtilerin hafifletilmesi için palyatif RT kullanımı önerilir.
İleri Evre AKK Tedavisinde Araştırılan Yeni Yaklaşımlar
Birçoğu moleküler hedefli tedavileri temsil eden, ilerlemiş AKK'nin tedavisi için birkaç yeni yaklaşım üzerinde çalışılmaktadır. Bu tedaviler yalnızca klinik deneyler bağlamında, genellikle hastalığın standart tedavilerde (örn. mitotan veya EDP-mitotan) ilerlemesinden sonra değerlendirilmiştir.
İmmünoterapi — Programlanmış hücre ölümü ligandı (PD-L1), bazı adrenokortikal karsinomlarda (AKK'ler) ve bunlarla ilişkili tümör infiltre eden lenfositlerde eksprese edilir ve bu hastalarda kontrol noktası inhibitörü immünoterapisinin etkinliği merak edilmektedir. Mikro satellit instabilite-yüksek (MSI-H) veya tümör mutasyon yükü-yüksek (TMB-H) durumları dışında immünoterapilerin AKK tedavisinde araştırılmasına ihtiyaç vardır.
Pembrolizumab, hem yetişkin hem de pediyatrik hasta grubunda yürütülen faz II çalışmalarında iyi bir güvenlik profili ve yüzde 14 ila 50 arasında değişen yanıt oranları göstermiştir:
- Açık etiketli, tek kollu bir faz II çalışmasında, ilerlemiş AKK'si olan 39 yetişkin hasta pembrolizumab ile tedavi edilmiştir. Çoğunluğu daha önce sistemik tedavi (ya mitotan ya da platin bazlı kemoterapi) almıştı. Yaklaşık 18 aylık ortanca takipte objektif yanıt oranı yüzde 23 (9 hasta) idi; ortanca progresyonsuz ve genel sağkalım sırasıyla 2 ve 24 aydı. Ek olarak, mikro satellit kararsızlığı yüksek/uyumsuzluk onarımı yetersiz (MSI-H/MMR-D) tümörleri olan altı hastanın ikisinde yanıtlar görüldü. Derece ≥3 toksisite oranları düşüktü (yüzde 13).
- Başka bir açık etiketli, tek kollu faz II çalışmasında da benzer sonuçlar görüldü. Bu çalışmada, önceki tedavilere dirençli AKK'li 16 yetişkin hasta (yaklaşık yarısında kortizol üreten tümörler vardı) programlanmış ölüm 1 (PD-1) inhibitörü pembrolizumab ile tedavi edildi. Yaklaşık yedi aylık takipte, iki hastada (yüzde 14) objektif yanıtlar ve yedi hastada (yüzde 50) stabil hastalık görüldü. Derece ≥3 toksisite oranları düşüktü (yüzde 13), sadece bir hasta pulmoner toksisite nedeniyle tedaviyi bıraktı.
- Çeşitli tekrarlamış solid (doku/organ kaynaklı) tümörleri olan 154 pediatrik hastayı kaydeden bir faz I-II açık etiketli pembrolizumab çalışmasında (KEYNOTE-051), adrenokortikal karsinomalı dört hastadan ikisi kısmi bir yanıt elde etti.
Diğer yaklaşımlar
IGF1R inhibitörleri – AKK'lerin yaklaşık yüzde 80'i, ağırlıklı olarak IGF-1 reseptörü (IGF1R) aracılığıyla sinyal verdiği bilinen insülin benzeri büyüme faktörü (IGF) tip 2'yi (IGF-2) aşırı ifade eder.
IFG1R'yi inhibe eden monoklonal antikorlar veya küçük moleküller ile ilgili preklinik ve erken faz çalışmaları başlangıçta ümit vericiyken, anti-IGF1R antikoru cixutumumabın müteakip bir faz I-II denemesi yalnızca sınırlı genel etkililik gösterdi. Ek olarak hem IGF1R hem de insülin reseptörünün oral küçük bir molekülü olan linsitinibin plasebo kontrollü bir faz III çalışması, ilerlemiş AKK'li hastalarda hastalıksız veya genel sağkalımda bir iyileşme göstermedi. Ancak, bu ilaçlarla tek ajan ve diğer ilaçlarla (temsirolimus gibi) kombinasyon halinde hastaların bir alt grubunda kısmi yanıtlar ve hastalık stabilizasyonu gözlendi.
İlerlemiş AKK'li hastalarda IGF1R inhibitörleri akla gelmesine rağmen, ileri çalışmalar hangi hastaların bu yaklaşımdan fayda görebileceğini öngörebilen moleküler belirteçleri belirleyebilir.
VEGF inhibitörleri – Vasküler endotelyal büyüme faktörü (VEGF), AKK tümör dokusunda yukarı regüle edilir ve bazı araştırmalar, dolaşımdaki VEGF düzeylerinin AKK'li hastalarda adrenal adenomlu hastalara kıyasla önemli ölçüde daha yüksek olduğunu bulmuştur.
Vaka raporları, antianjiyojenik ajanlar talidomid, sorafenib ve sunitinib aktivitesini öne sürerken, sonraki faz II denemeleri, VEGF inhibitörlerinin hem tek ajanlar (örn., aksitinib, sunitinib) veya kemoterapi ile kombinasyon halinde (örn., bevacizumab artı kapesitabin; sorafenib artı haftalık paklitaksel) sınırlı etkinlik göstermiştir.
EGFR inhibitörleri – AKK'lerin yüzde 80'inden fazlasının epidermal büyüme faktörü reseptörünü (EGFR) eksprese ettiği bulgusu, EGFR'yi hedefleyen ajanların incelenmesi için bir gerekçe sağlar. Ne yazık ki, gemsitabin ile kombinasyon halinde küçük moleküler EGFR inhibitörü erlotinib ile kurtarma tedavisi, diğer en az iki sistemik kemoterapi rejiminde başarısız olan ilerlemiş AKK'si olan 10 değerlendirilebilir hastadan oluşan bir çalışmada çok az fayda sağlamıştır. Sadece bir hasta küçük bir yanıt yaşarken, sekiz hasta hastalıkta ilerleme yaşadı.
Radyonüklid tedavisi – AKK, terapötik radyonüklidlerle de hedeflenebilir. Aktif radyonüklidlerin örnekleri arasında iyot-131-metomidat ve (R)-1-[1-(4-[131-I]iyodofenil)etil]-1H-imidazol- gibi 11-beta-hidroksilazı hedefleyen maddeler yer alır. 5-karboksilik asit azetidinil ([iyot-131]-IMAZA).
Hastalık Gidişatı Nasıldır?
Genel olarak, adrenokortikal karsinomda (AKK) hastalık gidişatı agresiftir ve 5 yıllık sağkalım oranları düşüktür. Beş yıllık sağkalım erken evre hastalıklar için yaklaşık %45 ila %60 ve ileri evre hastalıklar için %10 ila %25 arasındadır. Bunula birlikte AKK’de genel sağkalımın son yıllarda arttığını bildiren raporlar vardır.
- MD Anderson Kanser Merkezi'nde 20 yıllık bir dönem boyunca AKK tedavisi gören 139 yetişkin arasında, %34'ünün tanıda uzak metastazları olduğu gerçeğine rağmen, beş yıllık sağkalım oranı %60 idi.
- Memorial Sloan-Kettering Kanser Merkezi'nde tedavi edilen 113 hastanın ikinci serisinde, primer (yeniden tekrarlayan olmayan) hastalığın tam rezeksiyonu yapılanlar (68 hasta) %55 (ortanca 74 ay) beş yıllık sağkalıma sahipti.
Sonuç olarak adrenokortikal karsinomlar (AKK'ler), fonksiyonel olabilen ve Cushing sendromuna ve/veya virilizasyona neden olabilen veya fonksiyonel olmayan ve abdominal bir kitle veya tesadüfi bir bulgu olarak ortaya çıkan nadir, sıklıkla agresif tümörlerdir. Adrenal adenomların çoğu 4 cm'den küçüktür. Buna karşılık, keşfedildiğinde çoğu AKK'nin çapı 4 cm'den büyüktür. Kortikal adenomların lipid açısından zengin doğası, bilgisayarlı tomografi (BT) taramasında bu iyi huylu tümörü karsinomdan ayırmada yardımcı olur. Tanı anında, AKK'li yetişkin hastaların yaklaşık yüzde 50'si nispeten ileri evre hastalığa sahiptir.
1. Authors: André Lacroix, MDGary D Hammer, MD, PhD. Treatment of adrenocortical carcinoma. Literature review current through: Jan 2023. | This topic last updated: Aug 29, 2022. UpToDate.com
2. Author: André Lacroix, MD. Clinical presentation and evaluation of adrenocortical tumors. Literature review current through: Jan 2023. | This topic last updated: Apr 30, 2021. UpToDate.com